Featured
UCSC Genomics Institute sequences and assembles eleven human genomes in nine days




By Alexis Morgan
Communications Manager, UCSC Genomics Institute
May 06, 2020 — Santa Cruz, CA
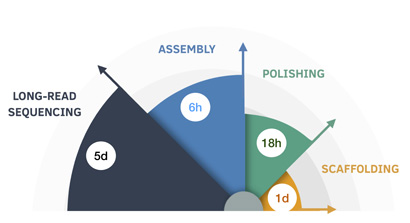
(Image above: The nine-day genome assembly process is broken down by length of time for each step. Credit: UCSC Genomics Institute)
The dawn of good, fast, and cheap human genome assembly has arrived, thanks in part to collaboration and innovation by an international team led by UC Santa Cruz researchers
It’s only been three years since UC Santa Cruz researchers proved that long-read human genome assembly using the same nanopore technology developed on campus could be done at all. At the time, it was a monumental effort, requiring 150,000 hours of computing time and weeks of work.
About a year later, using the PromethION nanopore sequencer, a similar effort proved significantly faster, cheaper, and easier, clocking in at about a week. “We sequenced eleven human genomes in nine days, which was unprecedented at the time,” said UC Santa Cruz Research Scientist Miten Jain.
Now, researchers at UC Santa Cruz have collaborated on an algorithm designed to accurately and precisely assemble individual, complete human genomes from long-read sequencing data in about six hours and for about $70.
The researchers said they hope their assembler will increase the pace of genomics research and open opportunities. This includes enabling pangenome research to represent the true scale of human diversity, a decidedly more practical pursuit.
Until recently, genomic research has relied exclusively on the reference genome from a single individual selected to represent an entire species. To reflect true human diversity, UC Santa Cruz has embarked on a pangenomic initiative to sequence 350 new, individual human genomes.
As part of this work, researchers at the UC Santa Cruz Genomics Institute developed a nanopore long-read sequencing protocol that consistently yields about 60X coverage (about 200 gigabases) of a human genome at unprecedented lengths (median read of 42 kilobases) using three PromethION flow cells. Additionally, about 7X coverage of the genome is in reads exceeding 100 kilobases in length. This method is highly scalable, both in terms of cost and the number of genomes that can be processed simultaneously. The team is now improving this method for higher read lengths and throughput, which will further facilitate the goal of achieving complete, phased, reference-quality genomes.
This large inflow of data necessitated the development of highly efficient software tools, starting with an assembler. “Our new assembler was designed to be cheap and quick, with the goal to be on the cloud,” said Benedict Paten, assistant professor of biomolecular engineering at UCSC. “It gives us the power to scale nanopore sequencing. Now, I’m confident that we’ll be easily assembling hundreds of de novo genomes in the next couple of years.”
An extensive team of researchers and developers that was led by Paolo Carnevali from the Chan Zuckerberg Initiative (CZI), and included many researchers at the Genomics Institute’s Computational Genomics Lab, contributed to this solution.
“When I saw Miten Jain’s 2018 paper, I was impressed and realized that I could contribute to the computational side of this line of investigation,” Carnevali said. “I had recently met Benedict Paten and decided I wanted to work with his team at UCSC.
The team were soon collaborating. Within months, they had developed and tested the special algorithmic sauce, which they called Shasta.
Shasta is an in-memory computing-driven algorithm that can now help complete a de novo (new, never before processed) human genome assembly in under six hours, the authors say, for an average cost of $70 per sample.
In a paper published May 4 in Nature Biotechnology, they describe how Shasta not only yields comparable or better accuracy as its contemporaries but also has the lowest number of misassemblies.
Not satisfied with this milestone, the team saw an opportunity to improve the draft assembly at an affordable cost and turn-around time. “To improve the base-level quality of the assemblies, we used a sequence polisher based on a deep neural network as the final assembly step,” explained lead author Kishwar Shafin, a graduate student in Paten’s group. “This brought the total cost of the assembly process to less than $200 and 37 hours, which further reduced the computational overhead of generating long-read assemblies dramatically, by a factor of five.”
The researchers assessed the precision and then validated the accuracy, and noted that they had achieved 99.9% accurate assembly using only nanopore data, a first for the human genome. Further, they generated chromosome-level scaffolds for these polished assemblies using HiC sequencing data.
Research scientist and co-author Karen Miga, who is directing the Data Production Center at UCSC for the Human Pangenome Project, points out the significance of the team’s achievements in improved accuracy.
“Our aim is not only to expand the diversity of the reference genome but also to resolve the hundreds of gaps that persist across the genome,” Miga said. “Now that we can routinely include these uncharted regions, we have a truly complete assembly of a human genome, and we can begin to explore variations of unknown consequence.”
###
Originally published here: https://news.ucsc.edu/2020/05/human-genome-sequencing.html
###
Tagged Genomics Institute, UC Santa Cruz



