Featured
Cancer study reveals powerful new system for classifying tumors




by Tim Stephen
UC Santa Cruz
One in ten cancers were reclassified in clinically meaningful ways based on molecular subtypes identified by a comprehensive analysis of data from thousands of patients
Cancers are classified primarily on the basis of where in the body the disease originates, as in lung cancer or breast cancer. According to a new study, however, one in ten cancer patients would be classified differently using a new classification system based on molecular subtypes instead of the current tissue-of-origin system. This reclassification could lead to different therapeutic options for those patients, scientists reported in a paper published August 7 in Cell.
“It’s only ten percent that were classified differently, but it matters a lot if you’re one of those patients,” said senior author Josh Stuart, a professor of biomolecular engineering at UC Santa Cruz.

Bioinformatics expert Joshua Stuart leads the Pan-Cancer Initiative. (Photo by Kevin Johnson/Sentinel)
Stuart helped organize the study as part of the Pan-Cancer Initiative of the Cancer Genome Atlas (TCGA) project. A large team of researchers from multiple institutions performed a comprehensive analysis of molecular data from thousands of patients representing 12 different types of cancer. This was the most comprehensive and diverse collection of tumors ever analyzed by systematic genomic methods. Each tumor type was characterized using six different “platforms” or methods of molecular analysis–mostly genomic platforms such as DNA and RNA sequencing, plus a protein expression analysis.
Cluster analysis
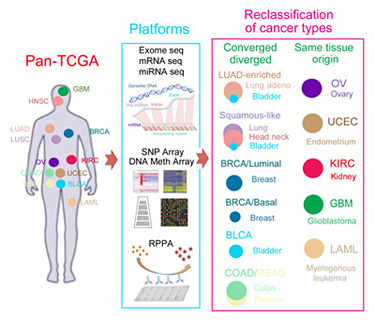
The research team used statistical analyses of the molecular data to divide the tumors into groups or “clusters,” first analyzing the data from each platform separately and then combining them in an integrated cross-platform analysis developed by co-first author Katherine Hoadley of the University of North Carolina. All six platforms as well as the integrated analysis converged on the same divisions of the cancers into 11 major subtypes. Five of those subtypes were nearly identical to their tissue-of-origin counterparts. But some tissue-of-origin categories split into several different molecular subtypes, and some subtypes encompass tumors with several different tissues of origin.
Bladder cancer was a particularly interesting group, because it split into seven different clusters, with most samples falling into one of three subtypes. One subtype was bladder cancer only, but some bladder cancers clustered with lung adenocarcinomas, and others with a subtype called ‘squamous-like’ that includes some lung cancers, some head-and-neck cancers, and some bladder cancers.
“If you look at survival rates, the bladder cancers that clustered with other tumor types had a worse prognosis. So this is not just an academic exercise,” Stuart said.
Other findings from the study reconfirmed cancer subtypes that were already recognized, such as the different subtypes of breast cancer based on well-characterized biomarkers. The findings provide a more refined, quantitative picture of the differences between breast cancer subtypes, Stuart said. For example, the results reinforce the idea that ‘basal-like’ breast cancers are a unique tumor type. “Basal-like breast cancers are as different from luminal breast cancers as they are from lung cancers,” he said.
The fact that all six platforms for molecular analysis identified the same set of subtypes, both individually and in multi-platform analyses, is an important result, Stuart noted. Not only does it give the researchers confidence in the subtypes they identified, it also means that different kinds of data can be used to classify a tumor.
Telltale signatures
“We can now say what the telltale signatures of the subtypes are, so you can classify a patient’s tumor just based on the gene expression data, or just based on mutation data, if that’s what you have,” Stuart said. “Having a molecular map like this could help get a patient into the right clinical trial.”
Although follow-up studies are needed to validate the findings, this new analysis lays the groundwork for classifying tumors into molecularly defined subtypes. The new classification scheme could be used to enroll patients in clinical trials and could lead to different treatment options based on molecular subtypes.
According to Stuart, the percentage of tumors that are reclassified based on molecular signatures is likely to grow as more samples and tumor types are included in the analysis (the next major Pan-Cancer analysis will include 21 tumor types). Coauthor Christopher Benz, an oncologist at the Buck Institute for Research on Aging and UC San Francisco, noted that the 10 percent reclassification rate in the current study is likely an underestimate due to the unequal representation of different tumors. “If our study had included as many bladder cancers as breast cancers, for example, we would have reclassified 30 percent,” Benz said.
The researchers reported that each molecular subtype may reflect tumors arising from distinct cell types. For example, the data showed a marked difference between cancers of epithelial and non-epithelial origins. “We think the subtypes reflect primarily the cell of origin. Another factor is the nature of the genomic lesion, and third is the microenvironment of the cell and how surrounding cells influence it,” Stuart said. “We are disentangling the signals from these different factors so we can gauge each one for its prognostic power.”
Data coordinator
The study involved an enormous amount of molecular and clinical data, which was managed by data coordinator Kyle Ellrott, a software developer in Stuart’s lab at UC Santa Cruz. The data sets and results have been made available to other researchers through the Synapse web site (www.synapse.org). Stuart worked with the bioinformatics company Sage Bionetworks to create Synapse as a data repository for the Pan-Cancer Initiative.
“It’s a huge amount of information, and all the data is available as programmable data sets that other researchers can use to do further analysis,” Stuart said. “The scale of this project is hard to imagine. All of the data that the TCGA project has been churning out got funneled into this paper, and it’s giving us an unbiased look at what the data have to tell us about cancer.”
The work was performed as part of the UCSC-Buck Institute Genome Data Analysis Center for the TCGA project led by Stuart, Benz, and David Haussler, director of the UC Santa Cruz Genomics Institute. The corresponding authors of the paper are Stuart, Benz, and Charles Perou of the University of North Carolina, Chapel Hill. The co-first authors are Katherine Hoadley of UNC; Christina Yau of the Buck Institute; Denise Wolf of UCSF; and Andrew Cherniack of the Broad Institute of Harvard and MIT. Stuart’s graduate students Sam Ng and Vladislav Uzunangelov also made significant contributions to the analysis.
This research was supported by grants from the National Institutes of Health. The TCGA project is led by the National Cancer Institute and the National Human Genome Research Institute. TCGA is a comprehensive and coordinated effort to accelerate our understanding of the molecular basis of cancer through the application of genome analysis technologies, including large-scale genome sequencing. The TCGA Pan-Cancer Initiative was launched in October 2012 at a meeting in Santa Cruz, California.
- Read original article here: http://news.ucsc.edu/2014/08/pan-cancer-study.html
###
Tagged UC Santa Cruz


